Neben der EU und USP versucht sich auch die WHO an einer Guideline, wie man mod-RNA Produkte auf Sauberkeit und Sicherheit hin prüfen könnte.

https://www.who.int/publications/m/item/annex-3-mRNA-vaccines-trs-no-1039
Das komplette Dokument ist im Konjunktiv geschrieben. Man sollte dieses und jenes tun. Das ist sehr schwammig und keine wirkliche, knallharte Guideline, die sehr viel Platz für Interpretation lässt. Anders als bei USP, wo klare, detailierte Analysevorgaben gemacht werden, welche Methode auf welche Art und Weise durchzuführen ist, gibt das WHO Regelwerkt generelle Problemzonen an, die die die WHO als zu kontrollieren ansieht.
Die Bereiche, welche die WHO in diesem Dokument anspricht, zeigen deutlich, welche Probleme man mitbekommen und als problematisch erkannt hat. Die WHO reiht sich dabei teilweise mit den Kritikern der modRNA-Plattform ein und würde von deutschen Medien wahrscheinlich als Schwurbler und Verschwörungstheoretiker bezeichnet werden.
SPOILER: Die WHO hat SEHR genau verfolgt, was die unabhängigen Forscher veröffentlicht haben.
Auch wenn alles sehr schwammig formuliert ist, ist der Leitfaden meiner Meinung nach der bisher beste Leitfaden. Deutlich besser als EU oder USP.
Wer hätte das gedacht. Die WHO meint es hier anscheinend ernst.
Einige der WHO Formulierungen und was geprüft werden müsste, sind vielleicht juristisch gut verwendbar. Die WHO ist in juristischem Kontext eine zitierfähige und belastbare Referenz, würde ich vermuten. Interessent ist vor allem, was noch nicht festgelegt ist, wie Grenzwerte und Messmethoden. Das zeigt, wo 2021 bis heute analytisch der Hund begraben ist, bei den aktuellen Produkten.
Dennoch gibt es immer noch das Schlupfloch für Notfälle, wo man es nicht so eng sieht und erneut neue Technologien in den Markt drücken könnte.
Ob die von der WHO geforderten Studien und Daten rückwirkend nachgeliefert werden müssen, wenn ein COVID Produkt dauerhaft auf dem Markt bleiben sollte, ist mir auch nicht ganz klar geworden.
6.2.1. mRNA sequence and arrangement of elements
Die annotierte Sequenz der DNA-Matrize sollte bereitgestellt werden.
Problem: muss diese vollständig annotiert sein? Pfizer hat den SV40 Promotor herausgenommen, der Rest der Matrize war beschriftet.
Auch die restlichen Vorgaben sind mehr als schwammig.
Sollten Sequenzen zudem gegenüber der nativen Sequenz verändert werden, um die Codons zu optimieren, sollten diese Änderungen beschrieben und begründet werden.
Jedes einzelne Codon oder generell, warum man optimiert hat?
Was ist mit Beschreibung der Änderung gemeint? Soll man den kompletten Effekt auf die 2D und 3D Struktur mit den damit einhergehenden potentiellen Änderungen bei der Proteinproduktion beschreiben (wie es sinnvoll wäre) oder reicht es einfach die Primärsequenz entsprechend zu listen und zu schreiben, dass man das für die Produktion so braucht?
6.2.2. Formulations and components
Chargenrezeptur: Die Chargenrezeptur für die kommerzielle Herstellung ist anzugeben. Die Mengen der einzelnen Bestandteile in einer einzelnen Impfstoffdosis sind aufzuführen. Das Gesamtvolumen einer Charge ist anzugeben. Wenn das Arzneimittel (Fertigimpfstoff) mehr als ein mRNA-Molekül (Wirkstoff) enthält, ist dies zu beschreiben, einschließlich der Angabe, ob die verschiedenen mRNA-Moleküle gleichzeitig in einem einzigen LNP eingekapselt oder getrennt in LNPs eingekapselt sind, die anschließend gemischt werden.
Wie genau müssen die Bestandteile angegeben sein, welche Range ist OK?
Eine Charakterisierung dieser Formulierungen, sowohl in chemischer Hinsicht als auch hinsichtlich der physikalischen Eigenschaften der strukturellen Formulierung (wie z. B. Nanopartikel), ist erforderlich und sollte Merkmale wie die Konsistenz und Stabilität der Formulierung und des Endprodukts abdecken. Auch Überlegungen zur Qualität der Lipide und zu den kritischen Qualitätsmerkmalen des Arzneimittels sollten einbezogen werden. Es sollte eine ausreichende Charakterisierung des mRNA-LNP und seiner Aufnahme in Zielzellen vorgelegt werden. Dies kann ein Verständnis der Oberflächenchemie, Größe, Polydispersität, Form, Ladung und Proteinbindungseigenschaften des resultierenden mRNA-LNP umfassen, um sicherzustellen, dass ein angemessener Schutz der mRNA und die erforderliche Stabilität des Impfstoffs erreicht werden. Wenn sich zeigt, dass die LNPs inhärente immunmodulatorische Wirkungen haben, sollten relevante Daten zu den potenziellen Vorteilen und Nachteilen vorgelegt werden. Daher sollten alle Eigenschaften der Formulierung, die sich auf die Sicherheit, Immunogenität und Wirksamkeit des Impfstoffs auswirken könnten, beschrieben und ihre Auswirkungen (positiv oder negativ) bei der Formulierungsentwicklung berücksichtigt werden.
Das klingt einerseits echt knackig, einige der Eigenschaften dürften schwierig zu bestimmen werden, andererseits wird das leider durch “ausreichende Charakterisierung” komplett entwertet. Wann und aus wessen Sicht muss die Charakterisierung ausreichend sein? Es geht hier nicht um Patientenschutz sondern nur um Stabilität. Die immunmodulatorischen Wirkungen werden erwähnt aber nicht definiert.
6.3.1 Quality of starting and raw materials and excipients
Die Ausgangsstoffe, Rohstoffe und Hilfsstoffe, einschließlich derjenigen, die zur Herstellung der mRNA verwendet werden (wie z. B. die DNA-Matrize, Nukleotide (die modifizierte Nukleoside enthalten können), Enzyme, Puffer, Lösungsmittel, etwaige Säulen zur Reinigung usw.) sowie die Lipide im LNP sollten beschrieben werden. Es sind Angaben zu Herkunft, Qualität, Kontrolle, Stabilität und Funktion dieser Stoffe zu machen, einschließlich der Angabe, an welcher Stelle im Herstellungsprozess die einzelnen Stoffe verwendet werden. Die Stoffe sollten für die Verwendung in der GMP-konformen Herstellung geeignet sein, und es sind Verweise auf international anerkannte Arzneibücher oder Einzelheiten zu ihren Spezifikationen anzugeben.
DAS ist aktuell nicht der Fall und könnte noch haarig werden, weil die Lipide Mischungen aus verschiedenen 3D Varianten sind, was hier nicht einmal thematisiert wird.
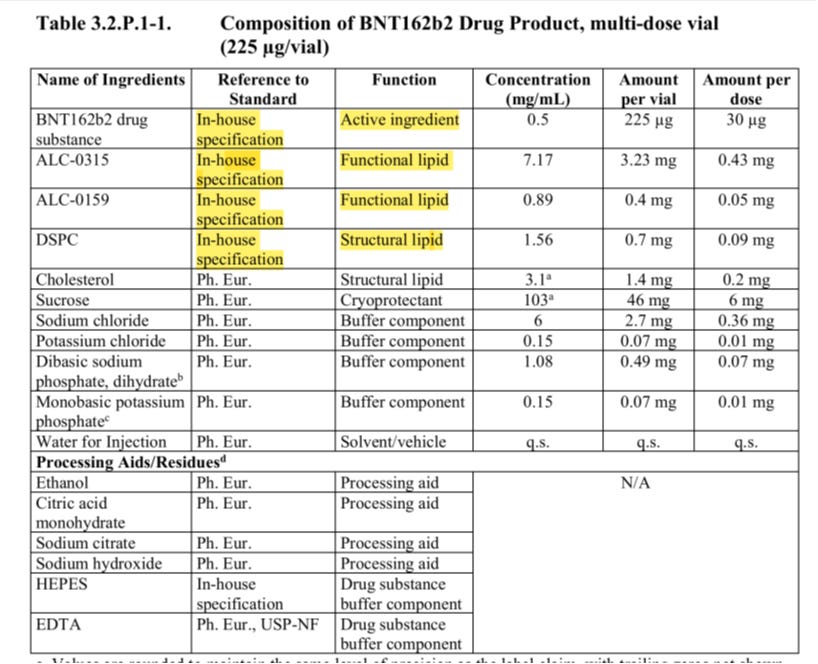
In Bezug auf die LNPs sollten die Herkunft und die Qualität der bei ihrer Herstellung verwendeten Lipide (insbesondere der in den LNPs enthaltenen neuartigen Lipide, die zuvor weder in nichtklinischen noch in klinischen Studien untersucht wurden) so detailliert beschrieben werden, dass eine aussagekräftige Bewertung ihrer Sicherheit und Qualität möglich ist. Auch für alle Hilfsstoffe, die nicht als neuartig gelten, sollten weiterhin geeignete Spezifikationen vorgelegt werden. Im Falle neuartiger Hilfsstoffe (z. B. kationischer Lipide) sollten, soweit möglich, Einzelheiten zum Herstellungsprozess und zur Kontrolle der neuartigen Lipide (einschließlich der Ausgangsstoffe und Zwischenprodukte) vorgelegt werden. Dies umfasst Informationen zu den vorgeschlagenen Ausgangsstoffen und etwaigen Zwischenprodukten, die bei der Synthese der neuartigen Hilfsstoffe verwendet werden, sowie eine entsprechende Begründung. Gegebenenfalls sollte die Durchführung einer Nitrosamin-Risikobewertung für die (kationischen) Lipide in Betracht gezogen werden.
Das ist wirklich nett. Weder USP noch EU haben die Lipide auf dem Schirm. Die WHO will eine Pharmacopeia Registrierung und klare Angaben über Herstellung und potentiellen Verunreinigungen.
Den Reinheitsgrad sollte man noch festlegen, das fehlt. Aber ich schätze, Pharmacopeia bedeutet 99,9% und das dürfte interessant werden.
Was die Sicherheit angeht, dazu gibt es aktuell gar keine Daten, die müssen erst erzeugt werden, auch das ist durchaus hilfreich.
Der Gehalt an Verunreinigungen, die mit den Hilfsstoffen in Zusammenhang stehen, sollte ebenfalls angemessen kontrolliert und begründet werden. Alle Reinigungs- und Isolierungsschritte sollten detailliert beschrieben werden.
Da ist die WHO sowohl USP als auch EU weit voraus, welche die Lipide komplett ignorieren.
Um die Qualität der vorgeschlagenen neuartigen Hilfsstoffe zu gewährleisten, sollte deren Hersteller auch über relevante Informationen zu den Analysemethoden verfügen, die für die Charakterisierung, die Stabilitätsüberwachung und die Chargenanalysen der Stoffe verwendet werden. Da die Einbeziehung eines PEGylierten Lipids eine entscheidende Rolle bei der Gewährleistung der In-vivo-Stabilität und der Verbesserung der zellulären Interaktion von LNPs spielt (42), sollten angemessene Kontrollen (beispielsweise von Molekulargewicht, Polydispersität und Molprozent) für das PEGylierte Lipid vorhanden sein. Bei der Herstellung von Hilfsstoffen wird die Einhaltung der WHO-Leitlinien für die gute Herstellungspraxis: Ergänzende Leitlinien für die Herstellung pharmazeutischer Hilfsstoffe (22) erwartet.
Der WHO ist also auch aufgefallen, dass man “vergessen” hat, die PEG-Länge zu definieren und festzulegen.
6.3.1.1 Quality of linear DNA derived from plasmid as starting material
Die Prüfung von DNA-Plasmiden (sofern diese zur Herstellung der linearen DNA verwendet wurden) und der linearen Matrize sollte Tests zur genetischen Identität mittels Sequenzierung, zur Integrität (einschließlich der Bestätigung der gewünschten kodierten Antigensequenz sowie der regulatorischen/kontrollierenden Sequenzen) und zum prozentualen Anteil an linearer DNA umfassen, sowie Tests (unter Verwendung geeigneter Referenzstandards) auf Restgehalte an genomischer DNA, RNA und Proteinen, auf Sterilität oder zulässige Keimbelastung sowie auf Endotoxingehalte.
Die WHO hat auf dem Schirm, dass BioNTech/Pfizer zu doof zum linearisieren waren. Ich bin beeindruckt.
6.3.2 Release of starting and raw materials and excipients
Wie bei jedem Impfstoff sollten für alle Rohstoffe Konformitätsbescheinigungen (sofern zutreffend) und Analysezertifikate vorgelegt werden, und es sollte eindeutig angegeben werden, welche Prüfungen vom mRNA-Hersteller durchgeführt werden oder ob das Material auf der Grundlage des vom Hersteller/Verkäufer/Lieferanten des Rohstoffs vorgelegten Analysezertifikats akzeptiert wird. Es sollte eine interne Richtlinie auf der Grundlage einer Risikoeinstufung nach Kritikalität für die hauseigenen Prüfungen und die Freigabe der im Herstellungsprozess verwendeten Rohstoffe festgelegt werden. Ausgangsstoffe sollten gemäß den Anforderungen und Spezifikationen für die Verwendung in der anschließenden GMP-Herstellung freigegeben werden.
Nichts mehr mit Firmengeheimnis, Analysezertifikate müssen für GMP transparent sein. DAS wird bei den Lipiden weh tun. So schwammig das auch formuliert ist, ohne exakte Vorgaben der Analysemethoden wie bei USP, dass hier die Analysezertifikate vorgelegt werden müssen und GMP konform sein müssen, dürfte teuer werden und weh tun. Ich vermute, bisher sind die Lipide eine schmutzige Angelegenheit.
6.4 Process development and in-process control
Die Validierung der Herstellungsprozesse sollte nachweisen, dass diese die kritischen und wesentlichen Parameter einhalten und ein Produkt liefern, das die vordefinierten Qualitätsmerkmale durchgängig erfüllt. Dies sollte den Nachweis umfassen, dass prozess- und produktbezogene Verunreinigungen reproduzierbar und konsistent auf Werte reduziert werden, die für die beabsichtigte Anwendung am Menschen akzeptabel sind.
Hier wären wir bei DNA Rückständen. Die Frage ist, wieviel ist akzeptabel?
6.5 Product characterization
Die Begründung für die Wahl der Analysemethoden zur Bestimmung verschiedener Eigenschaften sollte berücksichtigt werden, insbesondere wenn bei Verwendung alternativer Techniken – beispielsweise bei der Partikelgrößenmessung mit unterschiedlichen Methoden – wahrscheinlich ein anderes Ergebnis erzielt würde. Aus diesem Grund werden orthogonale Methoden empfohlen.
DAS klingt schwer danach, als wüsste die WHO, dass BioNTech/Pfizer immer exakt die Analysemethode genommen haben, die das Problem sicher NICHT detektiert und dass man das nicht so einfach durchgehen lassen will.
Es sollte nachgewiesen werden, dass das/die vollständige(n) kodierte(n) Protein(e) exprimiert wird/werden, ohne dass verkürzte oder alternative Formen vorliegen.
Das Problem verkürzter Proteine ist der WHO also bekannt.
Insbesondere wenn bei Charakterisierungsuntersuchungen die Expression verkürzter oder alternativer Formen des Zielantigens nachgewiesen wird und diese alternativen Formen zu Neoantigenen oder unerwünschten Immunreaktionen führen würden, kann dies eine Neugestaltung der mRNA-Sequenz erforderlich machen.
Die Probleme mit +1 Frameshift, längeren Varianten und unterschiedlicher Immunogenität von Varianten sind der WHO also auch aufgefallen.
Untersuchungen zur Partikelaufnahme könnten dazu beitragen, potenzielle Wirksamkeitsparameter zu charakterisieren, indem sie Aufschluss über die Zelltypen, die die Partikel aufnehmen, die Art oder den Mechanismus der Aufnahme sowie die Effizienz der Aufnahme geben, und somit als Orientierung bei der Auswahl der zellfreien oder In-vitro-Methode dienen, mit der sich diese Aktivitäten am besten bewerten lassen. Im Rahmen der Charakterisierung sollte ermittelt werden, ob eine dieser Eigenschaften als kritisches Qualitätsmerkmal und/oder als stabilitätsindikatives Merkmal kontrolliert werden sollte.
Das wird den Herstellern weh tun. Man weiß nur von 1-2% der LNP, dass sie transfieren, was der Rest so treibt ist unbekannt.
Die LNPs verteilen sich im ganzen Körper. In Zellkultur hat man gesehen, dass sie Expressionsergebnisse unterschiedlich sind.
Das zu normieren und Qualitätsstandards zu unterwerfen kann Jahre dauern, wenn das überhaupt kontrollierbar ist.
Bestimmte Aspekte der LNPs sollten sehr genau charakterisiert werden. Dazu gehört die Partikelgröße, die mittels verschiedener analytischer Verfahren bestimmt wird, um die morphologischen und dimensionalen Eigenschaften der mRNA-haltigen LNPs zu untersuchen.
Die Frage ist, wie eng der Rahmen in diesem Fall gesteckt wird. Aktuell ist er SEHR groß mit 40 – 180 nm

Die Messung der Oberflächenladung (zum Beispiel des Zetapotenzials) sollte ebenfalls als Methode zur Charakterisierung der LNPs in Betracht gezogen werden.
Das hat man bisher, wie es scheint, komplett vermieden. Die Partikel sollten eine Ladung von -3,13 mV haben. Wenn man das rigoros durchgesetzt hätte, wäre möglicherweise kaum eine Charge ausgeliefert worden. Auch dieses Problem ist der WHO also bekannt.
Mögliche Verunreinigungen, die durch die Ausgangsmaterialien eingebracht werden könnten, sowie mögliche produkt- oder verfahrensbedingte Verunreinigungen in der gereinigten mRNA sollten beschrieben und untersucht werden. Zu solchen Verunreinigungen können restliche bakterielle Wirtszellproteine (sofern diese zur Herstellung der DNA-Matrize verwendet wurden), Endotoxine, restliche bakterielle Wirtszell-RNA und chromosomale DNA (sofern Bakterien zur Herstellung der DNA-Matrize verwendet wurden), Enzyme (wie DNA- und RNA-Polymerasen sowie Restriktionsenzyme), nicht eingebaute Nukleotide, dsRNA, unvollständige oder unterschiedlich große RNA sowie andere im Herstellungsprozess verwendete Materialien gehören. Es sollten Daten zu den in der gereinigten mRNA vorhandenen Verunreinigungen vorgelegt werden, um die festgelegten Spezifikationen für deren maximal zulässige oder niedrigstmögliche Gehalte zu begründen. Für Verunreinigungen und Rückstände mit bekannten oder potenziellen toxischen Wirkungen wird erwartet, dass eine toxikologische Risikobewertung durchgeführt wird. Abgebaute mRNA kann im Rahmen von Analyseverfahren wie Polyacrylamid- oder Agarose-Gelelektrophorese, Hochleistungsflüssigkeitschromatographie (HPLC) und/oder Kapillargelelektrophorese bewertet werden. Der Grad der Konsistenz der Sequenz und Struktur der mRNA sowie deren Expression eines konsistenten Proteins bei der Transfektion in Zellen in vitro sind wichtige Merkmale, die für das Arzneimittel zu bestimmen sind.
Endotoxinwerte sind bisher in ALLEN Dokumenten geschwärzt. Da scheint es sein Problem zu geben. Auch dsRNA ist als Problem bekannt und DNA Rückstände sind nicht erwünscht. Die WHO kennt das Problem also. Bisher wird dsRNA eher nicht geprüft.
Etwaige Verunreinigungen (sowohl verfahrens- als auch produktbedingt), die aus den bei der Herstellung des Arzneimittels verwendeten Lipiden stammen könnten, sollten ebenfalls charakterisiert und untersucht werden. Dies ermöglicht eine Begründung der vorgeschlagenen Spezifikationsgrenzen, sodass diese Verunreinigungen angemessen kontrolliert werden und innerhalb des klinisch festgelegten akzeptablen Bereichs liegen.
Dafür muss man erst einmal die Toxizität und LD50 bestimmen. Das könnte interessant werden.
6.6 Consistency of manufacture
Wie bei anderen biologischen Arzneimitteln sollten vor der Beantragung der Zulassung mehrere aufeinanderfolgende Chargen unter Verwendung validierter Methoden getestet und analysiert werden, um die Herstellungskonsistenz zu bestimmen. Etwaige Abweichungen zwischen einzelnen Chargen, die außerhalb des akzeptierten Bereichs für die untersuchten Merkmale liegen, sollten vermerkt und untersucht werden. Die aus solchen Untersuchungen gewonnenen Daten sollten in Verbindung mit Produkt- und Prozesskenntnissen sowie der Bewertung der Kritikalität von Abweichungen bei bestimmten Merkmalen als Grundlage für die Begründung der gewählten Spezifikationen dienen. […]
In einigen Fällen kann jedoch bereits mit der Hochskalierung für die kommerzielle Herstellung begonnen werden, während die Zulassung für Material im Umfang einer klinischen Studie beantragt wird. Wann immer Änderungen am Herstellungsprozess vorgenommen werden, sollte die Vergleichbarkeit der Chargen nachgewiesen werden, insbesondere im Hinblick auf diejenigen, die in zulassungsrelevanten Studien verwendet wurden und nach dem vorgesehenen kommerziellen Verfahren hergestellt wurden. Protokolle zur Vergleichbarkeit und Strategien zum Nachweis der Vergleichbarkeit werden in den WHO-Leitlinien zu Verfahren und Datenanforderungen bei Änderungen an zugelassenen Impfstoffen (31) behandelt.
Das ist für die COVID modRNA nie passiert. Die 250 Probanden, welche das upscale Produkt bekamen, wurden offiziell nie ausgewertet.
6.7 Manufacture and control of bulk purified mRNA (drug substance)
Es sind Spezifikationen für kritische Qualitätsmerkmale hinsichtlich der Identität (siehe Abschnitt 6.7.1.1 unten), der Reinheit (Abschnitt 6.7.1.2), der Menge und des physikalischen Zustands (Abschnitt 6.7.1.3), der Sicherheit (Abschnitt 6.7.1.4) und der Qualität (Abschnitt 6.7.1.5) der gereinigten mRNA in Großmengen festzulegen und zu begründen. Es sind Beschreibungen der verwendeten Analysemethoden vorzulegen, die Akzeptanzgrenzen zu definieren und Informationen zur Validierung der Assays anzugeben. Die Ergebnisse der Prüfungen aller im kommerziellen Maßstab hergestellten Chargen sind zusammenzufassen und vorzulegen. Es sind außerdem Spezifikationen für die Stabilität unter Lagerbedingungen festzulegen.
Die Spezifikationen hat man leider bei den bisherigen Produkten nicht festgelegt, weil die Hersteller sich nicht einigen konnten, obwohl die WHO es 2020 versucht hatte.
“Das (WHO) Expertenkomitee für biologische Standardisierung (ECBS) hat Ende 2020 mehrere Sitzungen abgehalten, um regulatorische Überlegungen bei der Bewertung der Qualität, Sicherheit und Wirksamkeit von mRNA-basierten prophylaktischen Impfstoffen gegen Infektionskrankheiten zu dokumentieren. […] Der WHO-Ausschuss holte Input von Regulierungsbehörden aus allen WHO-Regionen sowie von Impfstoffentwicklern und -herstellern ein. […] Vertreter von BioNTech, CureVac und Moderna berichteten über ihre Erfahrungen mit der Herstellung, der Qualitätskontrolle sowie den nicht-klinischen und klinischen Aspekten bei der Entwicklung dieser klinischen Kandidaten. […]”
“Da die spezifischen Details der Herstellungsverfahren für Bulkware und Arzneimittel von Hersteller zu Hersteller unterschiedlich sind und viele dieser Informationen derzeit vertraulich behandelt werden, wurden die endgültigen Kriterien für die Zulassung den einzelnen nationalen Zulassungsbehörden überlassen. […] Obwohl die United States Pharmacopeia (USP) vor kurzem einen Entwurf für ein Kapitel über analytische Verfahren für die Qualität von mRNA-Impfstoffen mit Methoden zur Unterstützung der Prüfung von Qualitätsmerkmalen für mRNA-basierte Impfstoffe veröffentlicht hat, enthält das Dokument keine Akzeptanzkriterien. In Tabelle 2 sind die in der USP-Leitlinie vorgeschlagenen Analysemethoden zur Bewertung verschiedener Qualitätsmerkmale der gereinigten mRNA-Präparatsubstanz als Bulkware aufgeführt.”
“Es wurde auch festgestellt, dass der Gehalt und die Art der Verunreinigungen von Charge zu Charge stark variieren, insbesondere wenn sie in unterschiedlichen Größenordnungen und nach unterschiedlichen Verfahren hergestellt werden.”
Also nicht einmal die WHO, die angeblich die Weltherrschaft mit ihrem Pandemievertrag übernehmen will, war in der Lage, die Hersteller dazu zu bringen, Grenzwerte anzugeben und sich auf Akzeptanzkriterien zu einigen, OBWOHL USP schon einen Rahmen gesetzt hat, der von allen ignoriert wird. Dieses Regelwerk ist ein neuer Versuch der WHO, der zu begrüßen ist.
6.7.1.2 Purity and impurities
Jede Charge von gereinigter mRNA in Großmengen sollte auf ihre Reinheit geprüft werden, wobei das Ergebnis innerhalb der festgelegten zulässigen Grenzwerte liegen muss. Bei der Kontrolle der Verunreinigungen sollten auch die während der Herstellung eingebrachten Stoffe berücksichtigt werden, wie beispielsweise die DNA-Matrize, nicht eingebaute Nukleotide, nicht eingebaute Caps, Enzyme, mRNA-Fragmente und dsRNA.
Das dürfte interessant werden. Ob man sich auf Grenzwerte einigen kann? Geht dann überhaupt noch irgendeine Charge raus? Kann man diese Produkte überhaupt in einem Reinheitsgrad herstellen, wie er notwendig wäre?
Es ist wichtig, dass die Methoden zum Nachweis der Reinheit und zur Messung von Verunreinigungen auf einem möglichst breiten Spektrum physikalisch-chemischer, biologischer und/oder molekularer Eigenschaften beruhen.
6.8.1 Composition
Die endgültige Zusammensetzung des Impfstoffs, einschließlich des Wirkstoffs (mRNA) und aller Hilfsstoffe (z. B. Lipide), sollte zusammen mit der Menge der Bestandteile in jeder Darreichungsform beschrieben werden – insbesondere, wenn eine Zulassung für mehr als eine Dosierung oder Darreichungsform beantragt wird. Auch die Funktion der einzelnen Bestandteile sollte beschrieben werden.
Wie eng werden die Abweichungsbereiche dabei gesteckt?
Kennt man die Funktion jedes Bestandteils wirklich? Ist hier nur Chemie, Biochemie oder auch Immunologie inbegriffen?
6.8.4.6 Potency
Die Wirksamkeit jeder Charge des Endimpfstoffs sollte unter Verwendung geeigneter quantitativer und validierter funktioneller Methoden bestimmt werden. Zur Überprüfung verschiedener Aspekte der Wirksamkeit (einschließlich der Funktionalität) können unterschiedliche Tests erforderlich sein, die wahrscheinlich krankheitsspezifisch sind. Die Immunogenität beim Impfstoffempfänger ist eine komplexe Funktion der Eigenschaften des Endimpfstoffs, einschließlich der Verabreichung an Zielzellen durch seine Formulierung sowie der Expression der mRNA-kodierten Proteine (die eine sich selbst amplifizierende Replikonkomponente enthalten können). Daher können potenzielle In-vitro-Wirksamkeitstests zellbasierte Transfektionssysteme oder zellfreie Assays umfassen. Solche Methoden würden nachweisen, dass das Protein mit der richtigen Größe und Identität aus der mRNA exprimiert wird. Da die Wirksamkeit jedoch nicht nur auf der Grundlage des Produkttyps (in diesem Fall mRNA-Impfstoffe), sondern auch der klinischen Indikation der zu verhindernden Krankheit analysiert werden sollte, ist es nicht möglich, eine bestimmte Testmethode anzugeben, die zur Messung der Wirksamkeit verwendet werden sollte. Wie bei allen Qualitätskontrolltests sollte eine wissenschaftliche Begründung für die zur Kontrolle des Produkts ausgewählten Wirksamkeitstests vorgelegt und mit der klinischen Leistung in Zusammenhang gebracht werden.
Ich bin mir nicht sicher, ob die Wirksamkeit bei den bisherigen Produkte überhaupt je getestet wurde. Mir wurde gesagt, einige Batches der modRNA-Produkte hätten in Zellkultur teilweise nicht transfiziert, die Adenovirenprodukte hätten immer funktioniert und wären auch bei Raumtemperatur lange stabil.
6.10 Retained samples
Es sollte eine ausreichende Anzahl von Proben für künftige Studien und Anforderungen aufbewahrt werden. Zu diesen Anforderungen können unter anderem Produktionsuntersuchungen oder -entwicklungen, nichtklinische Studien oder künftige Brückenstudien gehören. Eine in einer zulassungsrelevanten klinischen Studie verwendete Impfstoffcharge kann als Referenzmaterial dienen; zu diesem Zweck sollte eine ausreichende Anzahl von Fläschchen zurückbehalten und ordnungsgemäß gelagert werden. Es ist eine vorausschauende Planung erforderlich, um die Aufbewahrung einer angemessenen Anzahl von Behältern der Charge aus der zulassungsrelevanten klinischen Studie zu ermöglichen.
Wurde das für die modRNA-Produkte gegen COVID gemacht?
7.2 Safety/toxicity in animal models
Die von der WHO sind wirklich üble Schwurbler. Was die alles von der armen Pharmaindustrie wollen:
a. Biodistribution und Persistenz: Der Aufbau einer Evidenzdatenbank zu diesem potenziellen Problem wird eine schnellere Entwicklung künftiger Impfstoffkandidaten ermöglichen (3, 66–71). Dieses potenzielle Problem könnte auch davon abhängen, ob der Impfstoff in bestimmte Zellen oder Gewebe wandert. Die NRA erwartet voraussichtlich nichtklinische Studien, die untersuchen, ob sich die mRNA und die LNPs (oder Lipidkomponenten) aus dem Gewebe, in das der Impfstoff verabreicht wurde, weiter verteilen, in welche Gewebe sie wandern und wie lange sie dort verbleiben. Eine Zustimmung zu diesen Studien sollte bei der NRA eingeholt werden.
Das waren lange geheime Daten, man behauptete, das Stoff bleibt an der Einstichstelle. Das sehen die Verschwörungstheoretiker der WHO anscheinend etwas anders.
b. Entzündung: RNA wirkt über verschiedene Wege entzündungsfördernd, insbesondere über das angeborene Immunsystem mit seinen zahlreichen Sensoren für RNA. Bei mRNA-Impfstoffen weisen sowohl die mRNA-Moleküle als auch die LNPs (die eine erfolgreiche Verabreichung und zelluläre Aufnahme ermöglichen) Eigenschaften auf, die das angeborene Immunsystem beeinflussen und auslösen können (72, 73). Auch wenn ein Teil dieser Aktivität für die Immunantwort auf den Impfstoff vorteilhaft sein mag, ist es wichtig, sowohl systemische als auch lokale Toxizität und Entzündungsreaktionen zu überwachen. Das Design nichtklinischer Studien muss alle Immunreaktionen, Reaktogenität oder Toxizitäten berücksichtigen, die auf Immunindikatoren (72, 73) für schwerwiegende unerwünschte Ereignisse oder unerwünschte Ereignisse von besonderem Interesse (AESI) beim Menschen hindeuten könnten. Darüber hinaus können andere zur Unterstützung der Verabreichung hinzugefügte Komponenten, wie z. B. PEG, obwohl sie relativ gut verträglich sind, ebenfalls die physikalisch-chemischen Eigenschaften und damit das Sicherheitsprofil beeinflussen (74–77). Daher ist es wichtig, das Gesamtprofil des Produkts einschließlich der Formulierung zu verstehen und zu wissen, wie physikalisch-chemische Eigenschaften (die variieren können) Entzündungen und das Sicherheitsprofil beeinflussen können. Die Wahl des Tiermodells wird wie immer entscheidend sein, wobei zu berücksichtigen ist, dass die angeborenen Immunantworten gegen RNA in Tiermodellen im Allgemeinen deutlich milder sind als die beim Menschen beobachteten.
Das klingt schwer nach Microarrayanalysen bei jedem Probanden. Das wird teuer.
Antikörper-Microarrays, Antigen-Microarrays, Peptid-Microarrays,Tissue-Microarrays (TMA), Genexpressionsanalyse, Protein-Microarrays… Was genau darf es den sein? Nichts davon wurde bisher gemacht.
c. Unerwartete und schwerwiegende Toxizitäten durch modifizierte Nukleoside: Einige antivirale Mittel und Krebsmedikamente, die spezifische unnatürliche Nukleosidanaloga mit veränderter Konformation enthielten, haben mitochondriale Toxizitäten verursacht, die zu Myopathie, Polyneuropathie, Laktatazidose, Lebersteatose, Pankreatitis, Lipodystrophie und sogar zum Tod führten. Einige dieser klinisch beobachteten Toxizitäten wurden jedoch in den verwendeten nichtklinischen Tiermodellen nicht beobachtet. Während die in den am weitesten fortgeschrittenen mRNA-Impfstoffen (gegen COVID-19) verwendeten modifizierten Nukleoside natürlich vorkommen, könnten zukünftige Impfstoffkandidaten Modifikationen enthalten, die unnatürlich sind. Daher muss insbesondere bei mRNA-Impfstoffen, die unnatürliche Nukleosidmodifikationen enthalten, die in anderen entwickelten nukleinsäurebasierten Produkten noch nicht gut charakterisiert wurden, sorgfältig geprüft werden, wie diese potenziellen Toxizitäten in geeigneten Tiermodellen und nichtklinischen Studien während der Sicherheitsbewertung beobachtet werden könnten (78–80).
Das hat man sich bei den COVID modRNAs komplett gespart.

d. Neuartige Lipide und neuartige LNPs: Da die zur Formulierung der LNPs verwendeten Lipide die Gesamtladung des Partikels beeinflussen, muss bei der Verwendung von LNPs, die mit neuartigen Lipiden hergestellt wurden, oder wenn die LNPs selbst modifiziert sind (z. B. durch veränderte Verhältnisse oder modifizierte Verfahren) und diese LNPs zuvor noch nicht in LNPs-verkapselten mRNA-Produkten nichtklinisch und klinisch getestet wurden, kann eine Bewertung der Toxizität der neuen Formulierung, die die neuartigen Lipide (oder etwaige neuartige Hilfsstoffe) enthält, erforderlich sein. Darüber hinaus kann die zuständige Behörde verlangen, dass die Genotoxizität und systemische Toxizität der neuartigen Lipidkomponente bewertet wird, ähnlich den Erwartungen für neuartige Adjuvanzien, die in den WHO-Leitlinien zur nichtklinischen Bewertung von Impfstoffadjuvanzien und adjuvierten Impfstoffen (19) und/oder für neue chemische Wirkstoffe in der ICH-Leitlinie S2 (R1) (62) dargelegt sind.
Diese Daten liegen für die modRNA-LNPs gegen COVID nicht vor. Muss man das nachholen?





It should be noted that early theoretical concerns during plasmid DNA vaccine development regarding the potential for integration of vaccine nucleic acids into the host genome do not apply to mRNA vaccines for the following reasons:
Der einzige bekannte Mechanismus, durch den sich RNA in das Wirtsgenom integrieren kann, setzt die Anwesenheit eines Komplexes voraus, der Reverse Transkriptase und Integrase enthält.
Menschliche Zellen, besonders von HIV-Patienten, haben reverse Transkripase.
Darüber hinaus sollte bei der Entwicklung von mRNA-Impfstoffkandidaten darauf geachtet werden, dass diese keine spezifischen RNA-Bindungsstellen für Primer enthalten, die die Reverse Transkriptase zur Einleitung der Transkription benötigt. Zudem müsste die RNA nach der reversen Transkription in den Zellkern transportiert werden, damit das entstandene Produkt integriert werden kann.
Daher sollten wohl eher keine SV40 Promotoren verwendet werden?
Schließlich wird die mRNA des Impfstoffs, sobald sie von den Körperzellen aufgenommen wurde, innerhalb relativ kurzer Zeit abgebaut, genau wie die körpereigene mRNA. Es wird davon ausgegangen, dass sich der mRNA-Impfstoff während dieser gesamten Zeit im Zytoplasma befindet, wo er translatiert und anschließend durch normale zelluläre Mechanismen abgebaut wird.
Da ist man bei der WHO wohl nicht ganz auf dem neuesten Stand. Die modRNA kann 28 Tage bis 15 Monate im Körper verweilen.
Liegen klinische Daten zu ähnlichen Impfstoffkandidaten vor, die auf derselben Plattformtechnologie basieren, sollte mit der zuständigen nationalen Behörde (NRA) geklärt werden, ob diese Daten wissenschaftlich ausreichend sind, um weitere nichtklinische Studien überflüssig zu machen.
Die Daten liegen zum Großteil nicht vor, müssten also noch erzeugt werden.
7.3 Accelerating nonclinical evaluation in the context of rapid vaccine development against a priority pathogen during a public health emergency
Sollte es erneut einen Notfall geben, kann man die alten Produkte, ohne die obigen Maßnahmen wieder in den Markt drücken. Da reichen so viel Daten wie in kurzer Zeit möglich erneut aus und GLP ist dabei nicht so wichtig.
Im Rahmen dieser Immunogenitäts- oder Schutzwirksamkeitsstudien sollten so viele Sicherheitsdaten wie möglich erhoben werden, wobei zu berücksichtigen ist, dass solche nichtklinischen Proof-of-Concept-Studien in der Regel nicht unter vollständiger Einhaltung der guten Laborpraxis (GLP) durchgeführt werden.
UND
Schließlich kann die nichtklinische Bewertung komplexer ausfallen und umfangreichere Studien erforderlich machen, wenn sowohl die LNPs als auch das kodierte Zielantigen (und damit die mRNA-Struktur und -Sequenz) neuartig sind; daher sollte auch eine Abstimmung mit der zuständigen nationalen Behörde erfolgen, und es ist möglicherweise nicht möglich, das nichtklinische Programm wesentlich zu verkürzen. Es könnte jedoch möglich sein, klinische Studien zu beginnen, während einige der erforderlichen nichtklinischen Studien parallel zur (oder etwas vor der) klinischen Entwicklung durchgeführt werden.
Teleskopierung ist im Notfall auch wieder OK.
Unterstützungsmöglichkeiten:
Bücherwunschzettel: https://www.amazon.de/registries/gl/owner-view/30LG3DJ4ET90L?ref_=list_d_gl_lfu_nav
Andere Unterstützungsmöglichkeiten für Holgers und meine Forschung:
Konto für Unterstützung für das Projekt Scan 2000
Dr. Merse DE34 4305 0001 0302 7851 75 Sparkasse Bochum
Horst Reissner: IBAN DE51 4401 0046 0406 4514 67
Dr. S. Stebel: https://ko-fi.com/einmalmitprofisarbeiten
Bitounis D, Jacquinet E, Rogers MA, Amiji MM. Strategies to reduce the risks of mRNA drug and vaccine toxicity. Nat Rev Drug Discov. 2024 Jan 23. doi: 10.1038/s41573-023-00859-3. Epub ahead of print. PMID: 38263456. https://pubmed.ncbi.nlm.nih.gov/38263456/
Priority-OCs-in-quality-LoQ-COVID-19-mRNA-Vaccine-BioNTech.docx S. 32 https://view.officeapps.live.com/op/view.aspx?src=https%3A%2F%2Fwww.covidtruths.co.uk%2Fwp-content%2Fuploads%2F2021%2F04%2FPriority-OCs-in-quality-LoQ-COVID-19-mRNA-Vaccine-BioNTech.docx&wdOrigin=BROWSELINK
Oude Blenke E, Örnskov E, Schöneich C, Nilsson GA, Volkin DB, Mastrobattista E, Almarsson Ö, Crommelin DJA. The Storage and In-Use Stability of mRNA Vaccines and Therapeutics: Not A Cold Case. J Pharm Sci. 2023 Feb;112(2):386-403. doi: 10.1016/j.xphs.2022.11.001. Epub 2022 Nov 16. PMID: 36351479; PMCID: PMC9637289. https://pubmed.ncbi.nlm.nih.gov/36351479/
Samaniego Castruita JA, Schneider UV, Mollerup S, Leineweber TD, Weis N, Bukh J, Pedersen MS, Westh H. SARS-CoV-2 spike mRNA vaccine sequences circulate in blood up to 28 days after COVID-19 vaccination. APMIS. 2023 Jan 17. doi: 10.1111/apm.13294. Epub ahead of print. PMID: 36647776. https://pubmed.ncbi.nlm.nih.gov/36647776/
Ota, N., Itani, M., Aoki, T., Sakurai, A., Fujisawa, T., Okada, Y., Noda, K., Arakawa, Y., Tokuda, S., & Tanikawa, R. (2025). Expression of SARS-CoV-2 spike protein in cerebral Arteries: Implications for hemorrhagic stroke Post-mRNA vaccination. Journal of Clinical Neuroscience, 136, 111223. https://doi.org/10.1016/j.jocn.2025.111223